|
PhD-SNP:
Predictor of human Deleterious Single Nucleotide Polymorphisms
PhD-SNP
is based a SVM-based classifier [1]. In the new version we developed a
predictor based on a single SVM trained and tested on
protein sequence and profile information (see figure below).
The PhD-SNP SVM input is build following the next steps:
-
for a given mutation the substitution form the wild-type residue
to the mutant is encoded in a 20 elemts vector that have -1 in the
position relative to the wild-type residue, 1 in the position relative
to the mutatnt residues and 0 in the remaining 18 positions.
-
a second 20 elements vector encoding for the sequience environment
is build reporting the occurrence of the residues in a windows of 19
residue around the mutated residue.
-
for a given protein, its sequence profile is built according
to the procedure detailed above. From this we evaluate
both the frequency of the wild type (Fi(WT)) and mutated
(Fi(MUT)) residues at position i. The NAL is the numeber is the number
of sequences in the alignment at a given and position and the
Conservation Index (CI).
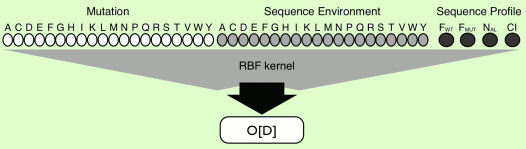
The
SVM-based method using sequence and Profile (SVM-Sequence)
The first SVM classifies mutations into diseases related (desired
output set to 0) and neutral polymorphism (desired output
set to 1). The decision threshold is set equal to 0.5. The
input vector consists of 40 values: the first 20 (the 20 residue
types) explicitly define the mutation by setting to -1 the
element corresponding to the wild type residue and to 1 the
newly intro-duced residue (all the remaining elements are
kept equal to 0). The last 20 input values encode for the
mutation sequence environment (again the 20 elements represent
the 20 residue types). Each input is provided with the number
of the encoded residue type, to be found inside a window centered
at the residue that undergoes the mutation and that symmetrically
spans the sequence to the left (N-terminus) and to the right
(C-terminus) with a length of 19 residues [2,3]. For SVM implementation
we use LIBSVM (http://www.csie.ntu.edu.tw/~cjlin/) with a
RBF kernel function K(xi,xj)=exp(-G ||xi -xj ||2)
The
SVM-based method using profile information (SVM-Profile)
The second SVM method (SVM-Profile) classifies mutations into
disease and neutral polymorphism taking as input only a vector
of 2 elements derived from the sequence profile. This is computed
from the output of the BLAST program [4], running on the uniref90
database (E-value threshold=10-9 , number of runs=1).
The first input element is the ratio between the frequencies
of wild-type versus that of the mutated residue in the sequence
mutated position and the second element is the number of aligned
sequences with respect to the mutation at hand. The software
and the kernel used for this SVM implementation are as described
above.
The
SVM-based method using sequence and profile information (PhD-SNP2.0)
The last vesion of PhD-SNP uses the same input described for the SVM-Sequence
method and 4 more profile based features. The sequence profile is calculated
accoriding to the procedure used for the SVM-Profile method but in this case
the input vector is composed by the frequenceies od wild-type and mutant
residues, the number of aligned sequences and the conservation index in the
mutated position
Results
The list of the predictions of PhD-SNP method are available on
OutPhD-SNP08.txt file.
In the table some scoring indexes of the efficiency of three
methods are listed.
|
Q2 |
P(D) |
Q(D) |
P(N) |
Q(N) |
C |
PhD-SNP |
0.76 |
0.76 |
0.72 |
0.76 |
0.80 |
0.52 |
SVM-Sequence |
0.68 |
0.68 |
0.64 |
0.69 |
0.72 |
0.36 |
SVM-Profile |
0.74 |
0.71 |
0.41 |
0.75 |
0.91 |
0.39 |
The overall accuracy Q2 is:
Q2=p/N
where p is the total number of correctly predicted residues
and N is the total number of residues.
The correlation coefficient C is defined as:
C(s)=[
p(s)n(s)-u(s)o(s) )] / D
where D is the normalization factor
D
=[(p(s)+u(s))(p(s)+o(s))(n(s)+u(s))(n(s)+o(s))]1/2
for each class s (D and N, for disease-related and neutral
polymorphism, respectively); p(s) and n(s) are the total number
of correct predictions and correctly rejected assignments,
respectively, and u(s) and o(s) are the numbers of under and
over predictions.
The coverage for
each discriminated structure s is evaluated as:
Q(s)=p(s)/[
p(s)+u(s)]
where p(s) and u(s) are as defined above. The probability
of correct predictions P(s) (or accuracy for s) is computed
as:
P(s)=p(s)
/ [p(s) + o(s)]
where p(s) and o(s) are previously defined (ranging from 1
to 0).
Required
Inputs
PhD-SNP is optimized to predict if a given sinle point protein
mutation can be classified as disease-related or as neutral
polymorphism. The required inputs are:
-
Protein
Sequence: the protein sequence can be provided
in raw format or giving its Swiss-Prot or uploading a
text file containing the protein sequence;
-
Position:
the position number in the sequence of the residue that
undergoes mutation;
- New
Residue: if you would ask for a specific mutation
please insert the symbol of the mutated residue;
-
Prediction:
choose between Sequence-Based or Sequence and Profile-Based
prediction.
-
Multi SVM:
choose if the prediction is performed using 20 different SVM model from cross validation procedure or a single SVM model (fast option).
The results can be sent to your e-mail address, if you ask
for it, or obtained interactively if you do not past your
e-mail in the proper box.
Outputs
The
output consists of a table listing the number of the mutated
position in the protein sequence, the wild-type residue, the
new residue and if the related mutataion is predicted as disease-related
(Disease) or as neutral polymorphism (Neutral).
The RI value (Reliability Index) is evaluated
from the output of the support vector machine O as
RI=20*abs(O-0.5).
The old help web page, where the datsets used in [1] are reported,
is reacheable with the following link.
[1] Capriotti, E., Calabrese, R., Casadio, R. (2006)
Predicting the insurgence of human genetic diseases associated
to single point protein mutations with support vector machines
and evolutionary information. Bioinformatics, 22:2729-2734.
[2] Capriotti, E., Fariselli, P., Calabrese, R. and Casadio,
R. (2005) Predicting protein stability changes from sequences
using support vector machines. Bioinformatics,
21 (Suppl 2), ii54-ii58.
[3] Capriotti, E., Fariselli, P. and Casadio, R. (2005)
I-Mutant2.0: predicting stability changes upon mutation
from the protein sequence or structure. Nucleic Acids
Res., 33 (Web server issue), W306-W310.
[4] Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang,
J., Zhang, Z., Miller, W. and Lipman, D. J. (1997) Gapped
BLAST and PSI-BLAST: a new generation of pro-tein database
search programs. Nucleic Acids Res., 25, 3389-3402.
|