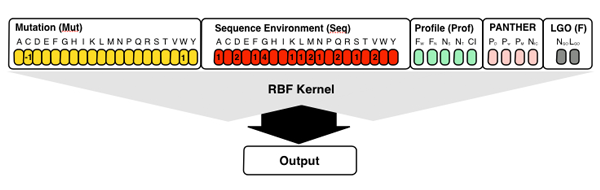
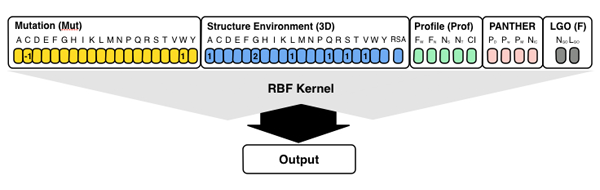
The server also implements S3D-PROF method that take in input different
subsets of SNPs&GO3d's input features. The S3D-PROD method takes in input the first 46
elements vector encoding for the structure and profile information. Selecting the
option "All methods" the predictions of S3D-PROF and standard sequence-based
SNPs&GO are calculated and included in the output.
Results
SNPs&GO was trained on a set of 38460 mutations and tested with a
cross-validation procedure over sets in which similar proteins were kept
in the same dataset also for the calculation of the LGO score, as derived
from the GO data base. At increasing input level of complexity, the
performance is also increasing, suggesting that on top of sequence
profile also LGO, derived from the protein GO annotation, is a crucial
added value for discriminating disease-related polymorphisms from neutral
ones. The finding that the level of performance increases at increasing
information added to the input corroborates the notion that support
vector machines can capture all the correlations existing in
complementary knowledge. Recently SNPs&GO was also tested by another
laboratory and scored among the best predictors available [6]. The
benchmark that we performed in house indicates that presently SNPs&GO is
one of the best scoring classifiers available for predicting whether a
mutation at the protein level is or is not disease-related.
Recently, SNPs&GO and SNPs&GO3d
have been tested on a dataset of 1,489 variants from 271
proteins with known structures (SAP-NEW).
Predictions have been obtained removing from the training set
all the variants from proteins with sequence identity higher
than 80% over more than 80% of the protein.
The efficiency of SNPs&GO and SNPs&GO3d
on SAP-NEW are compared with those of PhD-SNP and
and S3D-PROF methods.
|
Methods
|
Q2 |
P(D) |
Q(D) |
P(N) |
Q(N) |
MCC |
AUC |
|
PhD-SNP |
0.70 |
0.77 |
0.75 |
0.57 |
0.61 |
0.35 |
0.74 |
S3D-PROF |
0.74 |
0.77 |
0.84 |
0.65 |
0.55 |
0.41 |
0.78 |
SNPs&GO |
0.79 |
0.84 |
0.83 |
0.71 |
0.71 |
0.54 |
0.85 |
SNPs&GO3d |
0.84 |
0.84 |
0.89 |
0.85 |
0.78 |
0.68 |
0.91 |
|
The overall accuracy Q2 is:
Q2=p/N
where p is the total number of correctly predicted residues and N is the total number of residues.
The correlation coefficient MCC is defined as:
C(s)=[p(s)n(s)-u(s)o(s)] / W
where W is the normalization factor
W=[(p(s)+u(s))(p(s)+o(s))(n(s)+u(s))(n(s)+o(s))]1/2
for each class s (D and N, for disease-related and neutral polymorphism, respectively); p(s) and n(s) are the total number of correct predictions and correctly rejected assignments, respectively, and u(s) and o(s) are the numbers of under and over predictions.
The coverage for each discriminated structure s is evaluated as:
Q(s)=p(s)/[p(s)+u(s)]
where p(s) and u(s) are as defined above. The probability of correct predictions P(s) (or accuracy for s) is computed as:
P(s)=p(s) / [p(s) + o(s)]
where p(s) and o(s) are defined above (ranging from 1 to 0).
References
[1] Calabrese R, Capriotti E, Fariselli P, Martelli
PL, Casadio R. (2009). Functional annotations
improve the predictive score of human disease-related
mutations in proteins. Human Mutation. 30;1237-1244.
[2] Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z,
Miller W, Lipman DJ. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database
search programs. Nucleic Acids Res., 25, 3389-3402.
[3]
Thomas, P.D. and Kejariwal, A. (2004) Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A. 101:15398-15403.
[4] Capriotti E, Altman RB. (2011).
Improving the prediction of disease-related variants using
protein three-dimensional
structure. BMC Bioinformatics. 12 (Suppl 4): S3.
[5]
Kabsch, W. and Sander, C. (1983)
Dictionary of protein secondary structure: pattern
recognition of hydrogen-bonded and geometrical features.
Biopolymers.
22: 2577-2637.
[6] Thusberg, J., Olatubosun, A. and Vihinen, M. (2011) Performance of mutation pathogenicity prediction methods on missense variants. Human Mutation., 32, 358-368.